Abstract
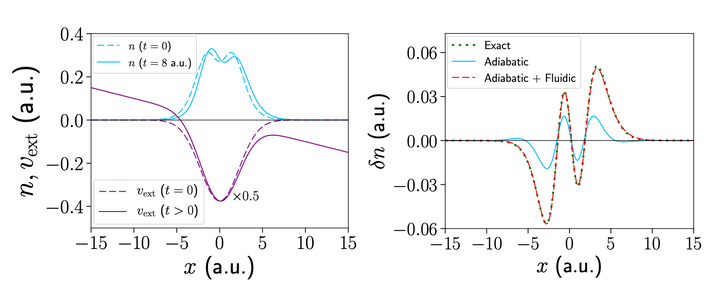
We present a simple geometrical “fluidic” approximation to the nonadiabatic part of the Kohn-Sham potential, vks, of time-dependent density-functional theory (DFT). This part of vks is often crucial, but most practical functionals utilize an adiabatic approach based on ground-state DFT, limiting their accuracy in many situations. For a variety of model systems, we calculate the exact time-dependent electron density and find that the fluidic approximation corrects a large part of the error arising from the “exact adiabatic” approach, even when the system is evolving far from adiabatically.
Type
Publication
Physical Review Materials