Abstract
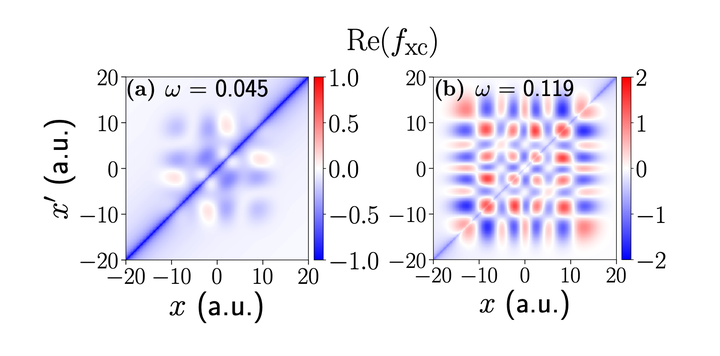
For two prototype systems, we calculate the exact exchange-correlation kernels fxc(x,x',w) of time-dependent density functional theory. fxc, the key quantity for optical absorption spectra of electronic systems, is normally subject to uncontrolled approximation. We find that, up to the first excitation energy, the exact fxc has weak frequency dependence and a simple, though nonlocal, spatial form. For higher excitations, the spatial behavior and frequency dependence become more complex. The accuracy of the underlying exchange-correlation potential is of crucial importance.
Type
Publication
Physical Review B